客服热线:
客服热线:


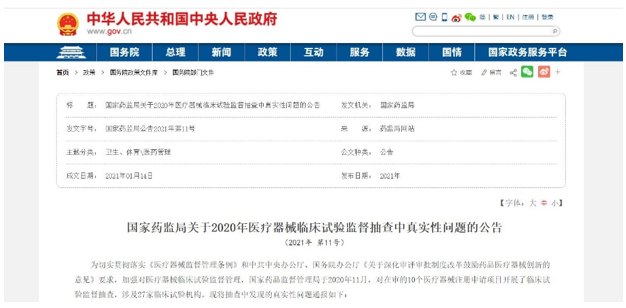
事实上,今年1月,国家药品监督管理局发布《关于2020年医疗器械临床试验监督抽查中的真实性问题的公告》,明确认定安旭生物的这一款监测试剂盒产品存在多项临床试验真实性问题,并根据《中华人民共和国行政许可法》和《体外诊断试剂注册管理办法》给予处罚。
自2015年以来,国家药品监管部门严格落实“四个最严”精神,颁布多项法规政策,旨在强化药械临床试验管理,重拳监管临床试验数据真实性、准确性、完整性问题,确保研究过程可追溯性。业内分析指出,如今的临床试验不应当抱有侥幸心理,真实、合规地开展临床试验是产品顺利获得监管认可的政策“红线”。
IPO前产品“爆雷”
临床数据无法溯源
为加强对医疗器械临床试验监督管理,国家药品监督管理局于2020年11月对在审的10个医疗器械注册申请项目开展了临床试验监督抽查,涉及27家临床试验机构。
国家药监局检查发现,杭州安旭生物科技股份有限公司生产的人类免疫缺陷病毒抗体/丙型肝炎病毒抗体/乙型肝炎病毒表面抗原/梅毒螺旋体抗体联合检测试剂盒(免疫层析法)(受理号:CSZ2000162)在浙江大学医学院附属第一医院开展的临床试验中,医疗机构留档的电子照片拍摄时间、地点与临床试验实际时间、地点不一致,临床试验数据无法溯源。
临床数据造假事件发生后,国家药监局根据《中华人民共和国行政许可法》和《体外诊断试剂注册管理办法》规定,对该注册申请项目做出不予注册决定,并自不予注册之日起一年内不予再次受理该项目的注册申请;同时,责成浙江省药品监督管理局切实履行对杭州安旭生物科技股份有限公司和相关临床试验机构的属地监管责任,依法依规调查处理。
值得注意的是,安旭生物是一家拟科创板上市公司。据悉,2020年6月2日,安旭生物科创板上市申请获受理,拟募资4.59亿元;2020年11月3日,安旭生物通过上市委会议;2020年12月30日,提交科创板注册。
安旭生物招股书显示,其主营POCT试剂及仪器,涵盖毒品检测系列、传染病检测系列等8大类,产品销售以外销为主,2017-2019年,安旭生物的境外收入分别为1.04亿元、1.54亿元、1.95亿元。
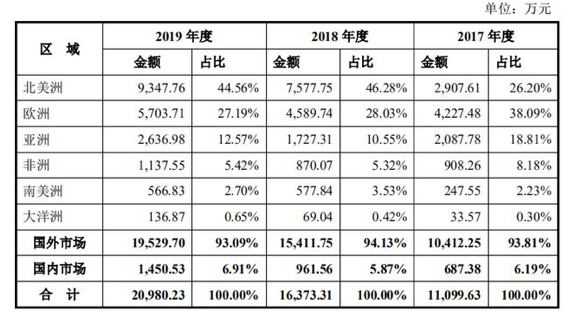
安旭生物招股书
事实上,安旭生物所处的细分市场竞争颇为激烈,境内主要对手包括东方生物、万孚生物、基蛋生物以及明德生物等。相比较而言,由于安旭生物在产品品种数量、生产规模及技术开发方面和头部企业仍存在较大差距,迫切需要通过资本市场助力企业发展。然而,就在企业上市关键时刻,重要在研产品爆雷,让行业对公司IPO未来前景不无担忧。
安旭生物此前公开表示,这次造假事件主要源自医疗机构出现问题,公司委托浙江大学医学院附属第一医院进行临床试验,抽查发现的问题主要责任对象是医疗机构,公司也是事件的受害者。
然而,这一说法显然无法平复质疑。市场分析认为,按照新版《药物临床试验质量管理规范》(GCP)要求,明确申办者作为临床试验质量和可靠性的最终责任主体,需要建立质量管理体系,基于风险进行质量管理,加强质量保证和质量控制,建立独立数据监查委员会,开展基于风险评估的监查和稽查等,企业责任在临床试验全周期质量管理中都是核心主体。
坚决贯彻“四个最严”
临床试验质量重拳监管
临床试验各主体建立完善的质量保证体系是确保项目高质量完成的基石。坚决贯彻“四个最严”精神,切实提高药械临床试验质量,促进临床试验水平的提升,强化临床试验研究者的责任意识,保证临床试验数据的真实性、准确性、完整性,确保研究过程可追溯性,正是药品监管部门多年来的工作重点。
3月10日,山西省药监局发布《关于开展对药物临床试验机构监督检查的通知》,决定对省内药物临床试验机构开展监督检查,旨在加强全省药物临床试验的监督管理,掌握药物临床试验机构开展药物临床试验的情况。
在此之前,黑龙江、山东、四川等省份陆续发布通知,加强药物临床试验机构备案管理工作,大幅提升临床试验监管水平;北京、天津、河北三地药品监督管理局共同制定的《京津冀药物临床试验机构备案后首次监督检查标准(征求意见稿)》,要求临床试验机构有不符合要求的“缺陷项目”立即整改。
临床专家指出,在真实性问题方面,参与临床试验的各方都应当敬畏生命、敬畏职责和规章,坚决不编造临床试验数据;确保临床试验数据可溯源;保证正确使用试验产品;不瞒报与试验产品相关的严重不良事件、产品缺陷、违禁治疗;确保注册申报资料中数据质量等。
在合规性问题方面,临床试验项目组成员应经过严格培训和授权,并熟练掌握相关法律法规和方案的具体要求,整个项目执行过程应有成熟的标准操作规程(SOP)作为指引。
为依法惩治药械注册申请材料造假的犯罪行为,顶层政策不断升级。最高人民法院、最高人民检察院于2017年发布《关于办理药品、医疗器械注册申请材料造假刑事案件适用法律若干问题的解释》明确:编造受试动物信息、受试者信息等药物非临床研究数据或者药物临床试验数据,影响药品安全性、有效性评价结果的,以“故意提供虚假证明文件”论处,最高可判五年。
前不久,中国裁判文书网公开了上海市普陀区人民法院的一则刑事判决书,爱恩康临床医学研究(北京)有限公司的一名员工,为加速推进百时美施贵宝(中国)投资有限公司委托其管理的项目,伪造了伦理审查机构的公章,违法用于向中国人类遗传资源管理办公室获取行政许可,最终因犯伪造事业单位印章罪获刑。
经过数年临床试验重拳监管,数据质量已经得到全产业链高度重视,监管部门对数据质量问题“零容忍”也得到了广泛赞誉。行业普遍认为,唯有真正夯实临床数据真实性、完整性、规范性,才可能铸就真正意义的药械科学研究和技术创新,切实保障上市药品的安全、有效和质量可控,从根本上提高中国制药工业的国际竞争力。