客服热线:
客服热线:

近两年正值药品注册法规调整年,无论是新法规后立项的新产品,还是现有的生产批文,研发实力不强的中小型生产企业都需要借助第三方——研发机构去完成项目。研发机构往往是完成中试放大后开始寻找买方,并且由于资本运作、品牌塑造等原因,研发机构在提交药物临床试验申请时往往选择申报自己为药品上市许可持有人,个别更是期望保持药品上市许可持有人的身份直到药品上市申请乃至批准阶段。但作为项目出资方,当然希望尽早获得上市许可持有人的身份。
而无论什么时候发起变更,都要涉及药品上市许可持有人变更。因此,变更相关政策中的细节需要仔细推敲而后行。此外,年底全面推行后,将利好哪些企业?对MAH项目感兴趣的接棒人,又需要具备哪些素质?
MAH制度试点方案 VS 药品注册管理办法修订稿(征求意见稿):
变更的三个现实问题
目前,对于药品上市许可持有人变更的政策,主要涉及两个:一是2016年6月6日发布的《药品上市许可持有人制度试点方案》;二是2017年10月23日发布的《药品注册管理办法(修订稿)》(征求意见稿)。
比较《药品上市许可持有人制度试点方案》和《药品注册管理办法(修订稿)》(征求意见稿)可以发现,《药品注册管理办法(修订稿)》(征求意见稿)对药品上市许可持有人和生产产地的变更等都有不少细节的变动(详见表1)。

1 研发机构就同一品种重复申请的界定
“多个主体联合只能一个主体作为药品上市许可申请的申请人,其他合作研发机构不得就同一品种使用相同资料重复申请”,这一规定出自《药品注册管理办法(修订稿)》(征求意见稿)。
而目前不少研发机构给多家生产企业研发同一个产品,在申报的时候资料会有相似性,那么资料是否相同的判断标准是什么?60%相似度还是90%的相似度?此外,目前国家对于相同产品相似配置合作研发机构的审批态度如何?会否采取“连坐制”,若其中一家合作研发机构的同一个产品出现问题则其它申请就算合作机构不同没有出现同类问题也可能会通不过?会否为了控制上市许可人的数量而减少同一品种同一合作研发机构所关联的药品上市许可持有人获批的数量?
2 药品上市许可持有人变更但受托生产企业和药品名称不变更,是否属于Ⅲ类变更?
相关的变更资料要求如何暂无文件模板细节。目前国内试点做的药品上市许可持有人变更更多的是集团内变更,暂无研发机构变更给生产企业的成功案例。在欧洲,由于药品上市许可持有人变更是指某药品上市许可持有人的法人(即实体)发生了改变,也就是说该产品的“上市许可”在两个不同的法律实体之间发生了移交或转让,但是不属于“药品上市许可变更”概念内,因此对其申请的处理由各欧盟成员国自行规定,即按照欧洲“成员国水平”来处理,申请的费用和申请程序的制定和管理都由各成员国自行设定,并没有统一的标准。
“药品上市许可持有人改变”的申请文件应当由当前的药品上市许可持有人(即原药品上市许可持有人)来递交。不过,新药品上市许可持有人也可以向药品评审委员会报告这一改变,此时,新药品上市许可持有人需要出示一份“委托书”,其内容要能表明原药品上市许可持有人同意新药品上市许可持有人递交“药品上市许可持有人改变”申请。
3 对过程变更规定的冲突
《药品上市许可持有人制度试点方案》要求在已受理药物临床试验申请或者药品上市申请、尚未批准阶段,申请人可以提交补充申请,变更申请人及受托生产企业。
但是,《药品注册管理办法(修订稿)》(征求意见稿)要求在药品上市许可申请审评审批期间,申请人原则上不得发生生产过程、质控和生产场地等方面影响产品质量、疗效、安全性的重大变更。此条可以理解为在申报过程中限制受托生产企业的变更。
MAH政策试点前后企业变化:
研发公司话语权大增
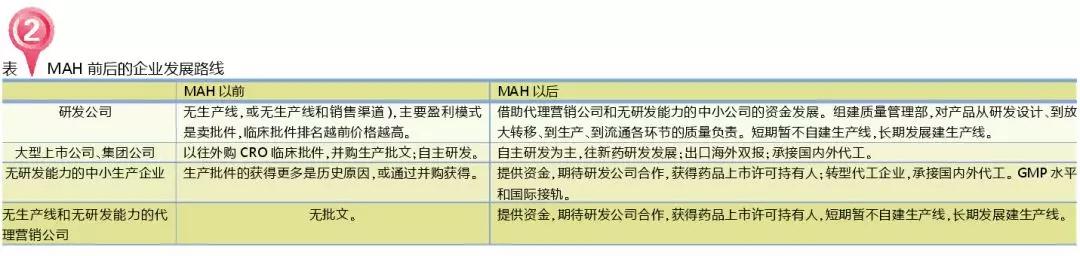
此外,笔者整理并总结了MAH制度试点前后对企业的影响(详见表2)。从目前的执行情况来看,《药品上市许可持有人制度试点方案》实施后,主要利好研发公司,它可以借助需要转型的无研发能力的代理营销公司和中小生产企业的资金。在一致性评价和药品再注册等法规的政策叠加效应之下,研发公司在技术转移的话语权大大提升。
新政策环境猜想:
变更“接棒者”需要哪些能力?
根据《药品注册管理办法(修订稿)》(征求意见稿),药品上市许可持有人对上市药品的安全性、有效性和质量可控性进行持续考察研究,履行药品的全生命周期管理,并承担法律责任。这意味着药品上市许可持有人需要建立药品全生命周期管理相关的质量管理体系及风险管理体系等(包括药物警戒管理系统)。也意味着药品上市许可持有人发生变更,药品的全生命周期管理责任要由新的药品上市许可持有人承接。
根据《药品上市许可持有人制度试点方案》政策解读,申请人(持有人)可以委托第三方主体开展药品质量监管工作,但相关委托不免除申请人(持有人)应当履行的义务与责任。也就是说,药品上市许可持有人变更、技术转移发生后,无研发能力的中小生产企业或代理营销公司仍可委托共同合作的研发公司开展药品质量监管工作。
目前来看,药品全生命周期管理相关的质量管理体系及风险管理体系等(包括药物警戒管理系统),最关键的还是要成立专业质量管理部门,要对生产企业的合规性审查(合同前和合作的过程中定期审计,GMP生产管理规范),生产过程的交批记录、质量分析报告等的审核、成品放行,药品上市后不良反应的监测、汇报、投诉、召回等事务的管理。还要加强与药监部门的沟通,不良反应、产品质量回顾、产品上市后的重大投诉和召回要定期汇报和沟通,委托生产厂的变更与产品上市后的改变注册标准的变更都要走审批程序,这需要该部门人员要承担一定的政务能力。