同时,为明确医疗机构备案应当具备的条件要求,为加强对高风险医疗器械管理,“备案管理办法”对备案的具体工作作了明确规定:
机构硬件——需对第三类医疗器械进行临床试验的,要为三级甲等医疗机构专业范围(科室);
人员硬件——具有高级技术职称并且参加过3个以上医疗器械或药物临床试验等;
备案程序——CFDA建立备案信息系统,相关医疗机构办理备案获得备案号后方可。
其实,CFDA于2015年4月便已开始就医械临床备案这一制度征求意见(《关于征求医疗器械临床试验备案有关事宜意见的函》(食药监械管便函〔2015〕25号))。此次发布的“备案管理办法”则是对中共中央办公厅和国务院办公厅关于医疗器械临床试验机构实行备案管理和取消“医疗器械临床试验资格认定” 要求的落实。
为保证临床试验工作的进一步开展,CFDA规定,自实施之日2018年1月1日到2018年12月31日为过渡期,自2019年1月1日起,医疗器械(包括体外诊断试剂)临床试验申办者应当选取已经在备案系统备案的医疗器械临床试验机构,按照有关要求开展临床试验。
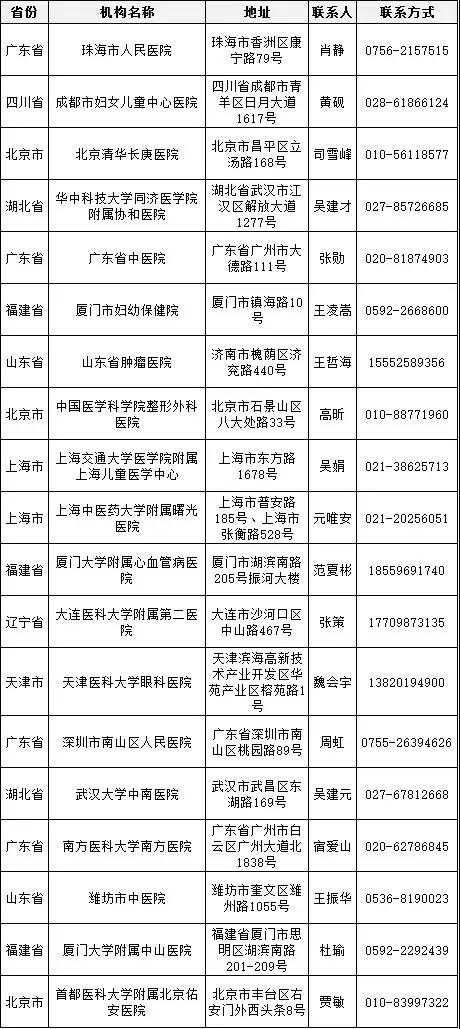
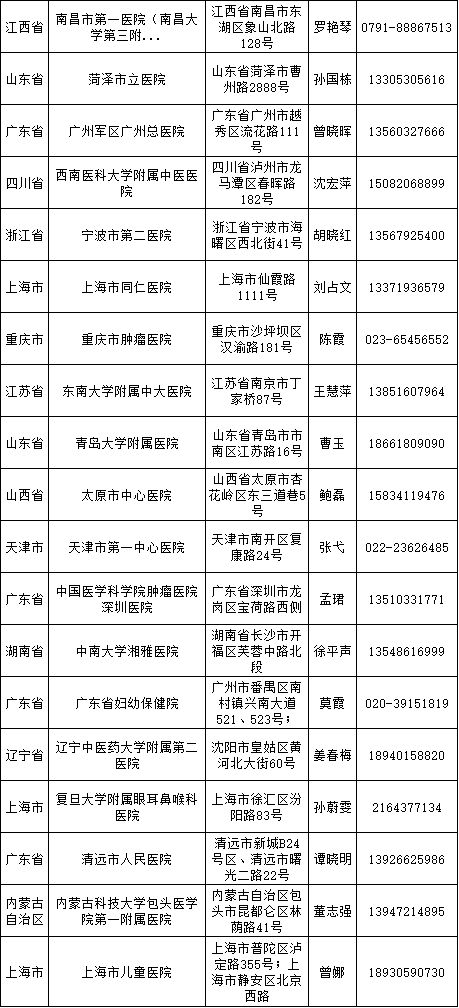
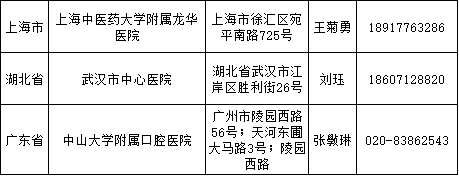
医疗器械临床试验逐步规范化,成功备案的医疗机构也在全国遍地开花。
截至2018年3月9日,已经成功备案医疗器械临床试验机构的医疗机构数量增加至41家,其中广东省最多,数目为9家、上海市6家、山东省4家、北京市3家、福建省3家、湖北省3家、天津市2家、四川省2家、辽宁省2家、江西省1家、浙江省1家、重庆市1家、江苏省1家、山西省1家、湖南省1家、内蒙古自治区1家。
临床专业科室涉及内科、外科、儿科、妇产科、影像科和检验科等。