而在国内,伴随着政策的支持、资本的青睐和大批技术人才归国创业,抗体类药物的研发和产业化也在近几年迅速崛起,技术水平不断提高,产业规模不断扩大。
研发水平的提高,加之海归人才的国际视野,让整个行业将从研发到销售各个环节的思维高度从国内逐步提升至全球。而向美国食品药品监督管理局(FDA)申请新药的临床研究申请(IND),是这一趋势的重要体现之一。
笔者就对到目前为止国内已成功申报美国IND的抗体类新药研发项目及其研发企业做一个汇总,并作简要分析,希望能借此以管窥豹,透视中国生物医药行业的发展现状和发展趋势。
【整体状况】
务实、差异化的国内研发布局
表1 国内药企申报美国IND成功的抗体类药物基本情况
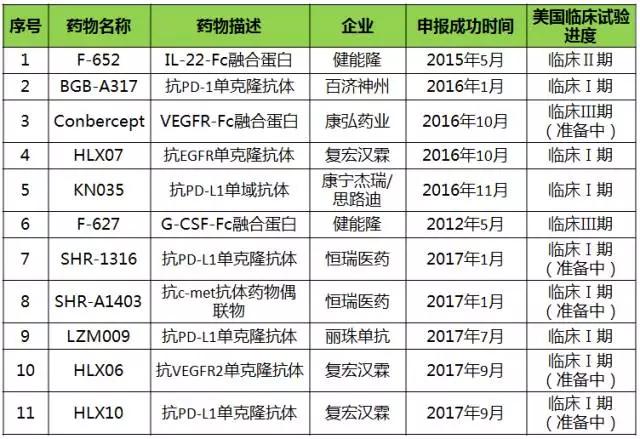
(注:美国临床试验进度截至2017年11月11日)
截至目前,笔者共收集到11款国内药企申报美国IND成功的抗体类药物(见表1)。
从企业来看,申报的企业既有恒瑞医药、康弘药业这样的转型的传统企业,也有康宁杰瑞、百济神州这样的初创公司,表明生物药(尤其是抗体类药物)的科学性和市场潜力已经得到国内医药界的普遍认同。
从所申报药物的靶点选择来看,都是较为热门的靶点,并没有明显的创新。但是,仔细分析可以发现,这些药物与已上市或在研的大部分同靶点药物相比,大部分还是做了一定程度的差异化。比如:
康宁杰瑞/思路迪的KN035的结构为单域抗体,这在已知的上市和临床在研同靶点抗体药物中,尚属唯一。经过验证,KN035能够常温保存,并且临床试验中的给药方式采用的是皮下注射,优势比较明显。
同是可治疗治湿性年龄相关黄斑变性的Fc融合蛋白,康弘药业的康柏西普(Conbercept)相比于再生元开发的Aflibercept,对融合蛋白受体部分的结构做了调整。
复宏汉霖的HLX06为抗VEGFR2单克隆抗体。相对于同靶点的上市单抗,礼来的Ramucizumab HLX06的抗原结合表位不同,在规避专利的同时,有望通过改变抗体的作用机制实现治疗效果的差异化。
百济神州的BGB-A317,则是在Fc段作了工程改造,消除了Fc段与其受体的结合能力,削弱Fc段引起的抗体依赖细胞吞噬作用(ADCP)效应,从而有望进一步降低抗体潜在的毒副作用;同时,抗体的耐高温和耐酸能力也得以提高。
可以看出,这些差异化策略,有些体现在靶蛋白结合位点的选择上,有些体现在药物结构的选择和改造上,最终的目标都是让药物获得一定的临床优势,确保其能够在激烈的竞争中占据一席之地,也就是我们所理解的“me-better”。这对于方兴未艾的国内生物医药行业来说,是一个比较务实的策略。
从临床申报的角度看,部分药物第一次临床申报时选择澳大利亚,比如百济神州的BGB-A317、健能隆的F-627,这是因为澳洲的临床申报时间较短、费用较低,并且临床数据可以获得美国和欧盟的认可。这对于研发竞争激烈的药物,或者研发经费相对紧张的初创企业,是个绝佳的选择。其他多数药物是逐步选择向FDA和国内CFDA同时申报,尤其是在中国已经加入ICH的大背景下,相信这种中美双报方式将成为国内企业未来的主要申报方式。
【亮点品种】
值得期待的创新项目详析
恒瑞SHR-A1403(抗c-met抗体药物偶联物)
看点: ADC产品,与全球其他c-met在研ADC项目比有技术优势
在11款国内药企申报美国IND成功的抗体类药物中,恒瑞的SHR-A1403是唯一一款抗体药物偶联物(ADC)。其靶点为酪氨酸激酶受体c-met,是一个在多种类型癌细胞表面高表达的生长因子受体,参与癌症的发生和恶化。
从目前c-met靶点的药物研发情况来看,失败率较高。小分子方面,在研项目较多,已上市的药物如Crizotinib等,均为多种酪氨酸激酶抑制剂,并非靶向性药物;抗体方面,还没有药物获批,普遍处于临床早期,进展最快的罗氏的Onartuzumab也在Ⅲ期折戟。分析可能的原因主要是:1)c-met这个靶点的激活机制较为复杂,简单地阻断它与配体的结合不一定能抑制它的活化;2)其下游的信号通路与其他靶点(如EGFR)下游的信号通路有交叉或代偿作用,这使得药物对它的抑制效果会被削弱。
因此整体看来,c-met靶点药物的单药治疗不是一个有效的手段。目前常用的改进方法包括与其他抗癌药物联合使用、研发双特异性抗体,以及开发ADC药物。
ADC药物的特点在于,这种治疗策略并不要求抗体发挥功能,而只是需要它能够特异性地结合抗原并引起细胞内吞,将小分子药物精确地带到肿瘤细胞内部并释放。因此,理论上来说,只要癌细胞高表达c-met,ADC药物就可以对其进行精确杀伤。这是一个非常巧妙的设计。目前,针对c-met这个靶点的在研ADC药物笔者已知的有3个,即艾伯维的ABBV-399、Sorrento的STI-0602和恒瑞的SHR-A1403。其中进度最快的是ABBV-399,目前处于Ⅰ期临床。
SHR-A1403抗体部分是利用抗原免疫小鼠制备杂交瘤,筛选阳性克隆,再通过人源化改造得来;毒素部分用的是恒瑞自主研发的新毒素。3个在研药物中,只有SHR-A1403采用的是定点偶联技术,这有助于控制药物的DAR(Drug Antibody Ratio)值和纯度。前期的功能实验证明,SHR-A1403能够有效杀伤特异性表达c-met蛋白的癌细胞系且效果明显强于单独的抗体,而对c-met不表达或低表达的细胞系则没有明显的杀伤作用;动物实验也证明了SHR-A1403能够有效抑制多种移植瘤的生长,且效果明显强于单独的抗体。
基于以上明显的功能和后续的一系列安全评价数据,以及CMC等其他必要的材料,恒瑞于去年12月向FDA提出SHR-A1403的临床试验申请。
今年1月份,恒瑞医药发布公告,其位于美国的控股孙公司Hengrui Therapeutics Inc.收到FDA的书面通知,可以开展SHR-A1403的药物临床试验。它是国内研发的第一个获FDA批准临床试验的ADC药物。
目前,从ClinicalTrials.gov上还无法查到SHR-A1403的临床试验信息,猜测该项目尚处在准备阶段。从在研药物的适应症来看,涵盖了多种癌症,并且包括了胃癌、肝癌等国人高发的癌症。因此,该药物若能够在临床上证明其安全性和有效性,其市场前景是比较广阔的。
健能隆F-627(贝格司亭,G-CSF-Fc融合蛋白)
看点:第三代产品,但长效G-CSF类竞争激烈,Ⅲ期临床结果是关键
G-CSF(granulocyte colony-stimulating factor,粒细胞集落刺激因子),被认为是促进中性粒细胞发育和从骨髓向外周释放的关键因子,能够通过激活其受体调控中性粒细胞的早期发育、存活、迁移和活化。因此,G-CSF理论上可用于治疗各种原因导致的中性粒细胞缺失。
而F-627就是一款重组人G-CSF-Fc融合蛋白,有望用于化疗引起的嗜中性粒细胞减少症的治疗。它利用了健能隆的双分子技术平台,将人源的G-CSF分子与抗体的Fc段(IgG2亚型)相连。由于Fc段可以通过链间二硫键形成二聚体,因此一个药物分子里包含两个G-CSF分子。
实际上,重组G-CSF蛋白并不是一类新颖药靶的药物。早在1991年,FDA就批准了安进的Filgrastim,即第一代重组人G-CSF,用于中性粒细胞减少症的治疗。由于分子的体内半衰期较短,因此需要每次化疗后多次注射。之后,安进对Filgrastim进行聚乙二醇化(PEG)修饰,得到第二代重组人G-CSF(pegfilgrastim,Neulasta)(于2002年上市,2016年全球销售额46亿美元),其半衰期得以延长,每次化疗后仅需一次注射,但与G-CSFR的亲和力由于N端的PEG修饰而降低。目前在研的长效重组人G-CSF(包括Sandoz和Coherus的产品)都是第二代的仿制型。而健能隆的F-627则属于第三代。融合的Fc的作用除了延长分子的半衰期之外,还由于携带两个G-CSF而能够促进G-CSFR的二聚化,从而增强对下游信号的激活作用。
2011年于澳大利亚完成的Ⅰ期临床试验中,F-627显示了良好的安全性和药代动力学特点;国际多中心Ⅱ期临床试验(美国的临床试验于2012年5月获FDA批准,整个试验于2014年10月结束)的数据显示,F-627的治疗效果不劣于对照药Neulasta,半衰期比对照药略长。
国际Ⅲ期临床申报于2016年5月得到FDA批准。该试验由两部分构成:GC-627-04和GC-627-05。GC-627-04是第一阶段,已于去年12月启动,是将F-627与安慰剂进行对照的一项随机、多中心、双盲试验,评估F-627的安全性与有效性,目前已在美国和欧洲多个国家完成患者入组。GC-627-05是第二阶段,是对比F-627与对照药Neulasta的安全性和有效性的随机、多中心、开放标签试验。10月10日,健能隆收到FDA回复,FDA通过“特殊方案评估”(Special Protocol Assessment,SPA)程序,与健能隆就GC-627-05方案达成具有约束力的协议,同意其使用“特殊方案评估”开展Ⅲ期临床试验。
“特殊方案评估”是FDA建立的一个对在研药物临床试验方案进行提前探讨和确定的程序,探讨内容包括临床试验方案的设计、入组病人的筛选、主要和次要终点的判断、临床数据统计分析等试验设计的各个环节,这些环节相关细节的确定将有助于未来的临床试验满足新药获批所需的监管要求。新药研发单位与FDA就上述计划达成一致性协议后,“特殊方案评估”的程序获得通过,新药研发单位将按照FDA批准的临床方案进行试验。这有助于提高新药的上市申请的成功率。因此,对于F-627的临床试验及未来的上市申请,我们应该给予更多的期待。
目前的长效G-CSF类药物市场竞争较为激烈,国内外有多家企业的产品已上市或在研。若想占据一定的市场份额,F-627必须在Ⅲ期临床表现出色,尤其是在GC-627-05方案中,需要证明自己不劣于甚至明显优于对照药;在上市后,还要注重学术推广和销售渠道的建设。
健能隆F-652(普罗纳亭,IL-22-Fc融合蛋白)
看点:全球首创生物药,在同类药物和疾病市场的竞争中均占优
F-652是健能隆的另一款细胞因子融合蛋白药物,是将重组人白介素-22(IL-22)与抗体Fc段融合表达。
IL-22的功能较为广泛,在多个器官的功能维持、损伤修复等过程中扮演重要的角色。因此,F-652作为重组的IL-22,有望用于治疗多种器官损伤和炎症疾病。
F-652于2013年在澳大利亚完成了以健康志愿者为受试者的F-652临床Ⅰ期试验。结果显示,F-652具有良好的安全性、预期的药代动力学和诱导生物标志物的特性。目前,在澳大利亚和中国开展的药物安全性和药代方面的评价工作已经完成,另有一个Ⅰ期试验计划在中国开展,用于检测该药物在急性胰腺炎患者体内的药效。2015年5月,F-652获FDA批准临床试验。目前两个Ⅱ期临床试验已经在美国进行,分别是:NCT02655510,健能隆与美国梅奥医院合作,评估药物在急性酒精性肝炎患者体内的安全性和有效性;NCT02406651,健能隆与美国纪念斯隆-凯特琳癌症中心合作,F-652与系统性的系统性皮质类固醇激素(Systemic Corticosteroids)联合,用于接受造血干细胞移植的患者新近诊断的伴发Ⅱ~Ⅳ级下消化道症状急性移植抗宿主病的治疗。
F-652是健能隆具有自主知识产权的全球首创生物药(first-in-class)。从笔者的搜索结果来看,目前临床在研的重组IL-22除了F-652,只有罗氏的RG-7880,处在Ⅰ期临床,所选的适应症为溃疡性结肠炎和糖尿病足溃疡。因此,从同类型药物的竞争情况来看,F-652的增长空间很大;放置于整个疾病市场的竞争中,F-652可以凭借其良好的靶向性、靶点特异性和体内药代特征,在特定的细分市场拥有竞争力。基于器官损伤、炎症这些类型疾病的巨大患者基数,这一类的药物的市场前景值得期待。
结语>>>
笔者认为,国内目前涉足抗体类药物研发的企业按性质大致可分为3类:一是由风险资本支撑的初创企业,如康宁杰瑞等;二是传统企业与新技术、与海归人才合作成立的子公司,如丽珠单抗和复宏汉霖等;三是处在转型中的传统企业(包括内部研发和外部股权投资/并购),如恒瑞医药、天士力。目前申报IND成功的国内企业,囊括了以上三种类型,显示国内医药行业对抗体类药物技术和市场的普遍重视。
当然,国内的抗体药物行业尚处在起步阶段,目前的靶点选择基本集中在少数热门靶点上。在早期,这样的策略是可行的,因为国内市场需求由于支付能力和进口药物审批速度的制约而远未得到满足。开发同靶点药物和生物类似药风险更小,理论上可以降低成本从而降低药价,惠及国内患者。
但是,随着中国加入ICH以及中国对于进口药品审评速度的大幅增加,目前这种相对低层次的研发很难持续。首先,在拥有患者比例(共性)和支付能力(特性)两个天花板的国内医药行业中,这样的扎堆研发免不了让人有产能过剩的担忧;而且中国作为全球第二大经济体,整个行业停留在“me-too”或者极其有限的创新程度上,也必然受人诟病。
不过整体环境正在变好,政府的多项政策措施在为新药研发扫除障碍,企业和资本市场对新药研发投入更多,海外人才一批批归国。这一切,都预示着整个行业和抗体药物产业将会有更好的发展。
笔者认为,在未来,抗体药物领域需要在如下几个方面持续努力:第一,理性进行同靶点药物和生物类似药的研发,通过合理地降低成本来让患者受惠;第二,注重靶点和技术创新,在为企业寻求生存之道的同时,开拓药物能够覆盖的疾病领域,寻求有效的差异化,让更多的疾病有药可治,让更多患者的生活质量有所提高。
我们相信中国抗体药的明天会更好!